首页 / 产品和服务 / 产品类型 / 抗体产品 / 特色抗体推荐
Maintaining normal metabolism balance is critical to ensure all life activities smooth. There are two major catabolism pathway of eukaryotic cells: the ubiquitin proteasome pathway and the autophagy pathway[1]. The ubiquitin proteasome pathway is characterized by the ubiquitination of the proteins, and degradation of the proteins by proteasomes finally. This way mainly selects the degradation of short-acting intracellular proteins, while the intracellular long-acting macromolecules and impaired organelles are mainly degradated through the autophagy way. Many studies have shown that the normality of autophagy plays an very important role in the stability of the intracellular environment. The autophagy barriers seriously impair the normal differentiation of embryonic cells and embryonic development in the process of development. Moreover, autophagy is also closely associated with the occurrence and development of many diseases, such as neurodegenerative diseases, cancer, infection of pathogenic microorganism and so on[2].
Autophagy is an evolutionarily conserved cellular degradation pathway that involves breakdown of cytoplasmic components by the lysosome. Although “autopahgy” was described in the late 1950s, its mechanism was elucidated much later using yeast genetics. Expectedly, the discovery of autophagy has won the Noble Price. Because it provided a new sight into the normal physiologically process and a new target for many disease therapy. Autophagosome formation requires three main steps: initiation, nucleation, and expansion. In general, autophagy is categorized by three main types: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy[3, 4].
(1)Microautophagy
Microautophagy is a non-selective lysosomal degradative process which directly engulfs the cytoplasmic cargo and eliminates them by both invagination and vesicle scission[5]. While microautophagy is unresponsive to amino-acid deprivation[6], and the mechanisms about microautophagy regulation is still known little.
(2)Chaperone-mediate autophagy(CMA)
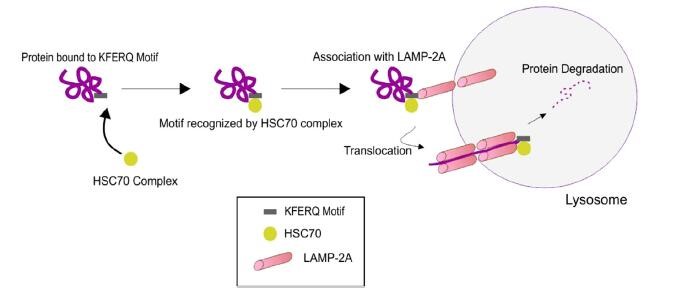
During CMA, cytoplasmic cargo is targeted to the lysosome, where it is degraded by lysosomal enzymes[7](Figure 1). The pentapeptide motif of CMA substrates contains a glutamine (Q) residue at the beginning or end of the sequence, one or two of the positive charged amino acids lysine(K) or arginine(R), one of the hydrophobic amino acids, phenylalanine(F), valine(V), leucine (L) or isoleucine(I) and one of the negatively charged amino acids, glutamic (E)or aspartic acid(D)[7]. In the cytosol, a constitutive chaperone, heat shock cognate protein of 70kDa(Hsc70), along with other co-chaperones(Bag1, Hip, Hop and Hsp40), bind to the substrate on the pentapeptide motif KFERQ, which is present in the amino acid sequence of all CMA substrates, consequently transporting it to the surface of the lysosomal membrane[8].
Once the substrate complex is targeted to the lysosomal surface, it interacts with the cytosolic tail of lysosomal-associated membrane protein type 2A(LAMP-2A).The monomeric LAMP-2A forms multi-protein complex structures, along with many other proteins, promoting the translocation of CMA substrates.CMA substrates can be introduced to this multi-protein complex in the folded or unfolded state; however, translocation of the substrates can only be carried out in the unfolded form[9]. The folding and unfolding of CMA substrates are tightly regulated by Hsc70 and the other co-chaperones. Once the CMA substrates are internalized into the lysosomes, they are degraded by lysosomal hydrolases. Subsequently, LAMP-2A dissociates from the multi-protein complex to form monomers, where another CMA substrate can bind, and thus this dynamic process maintains the homeostasis of CMA [10]. Alterations in redox balance and subsequent oxidative stress is one of the major factors that regulate the levels of LAMP-2A[10]. The role of CMA in skeletal muscle has not been widely studied. Increased LAMP2A has been reported in mouse skeletal muscle after a single bout of exercise[11]. Additionally, abnormalities of CMA have been observed in sporadic inclusion-body myositis muscle fibers[12].
(3)Macroautophagy
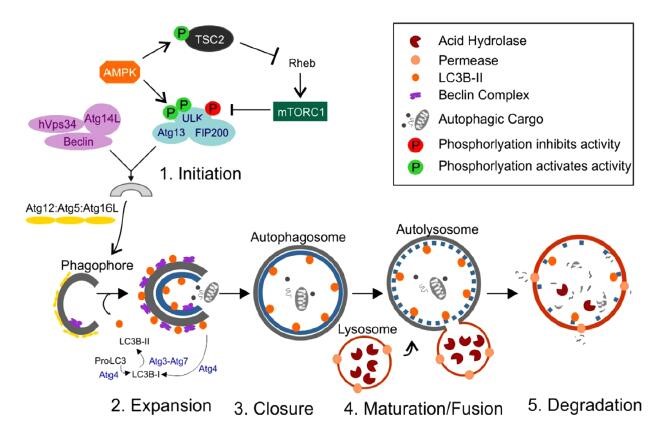
Hypoxia, extracellular pH changes, nutrient deprivation, oxidative and endoplasmic reticulum stress are signals for autophagy in cancer cells. Autophagy is a homeostatic mechanism, that, when disrupted, can promote and accelerate tumorigenesis. The autophagic process is a highly regulated process that has been divided into five stages: induction, vesicle nucleation, vesicle elongation, docking and fusion, and degradation and recycling (Figure 3).
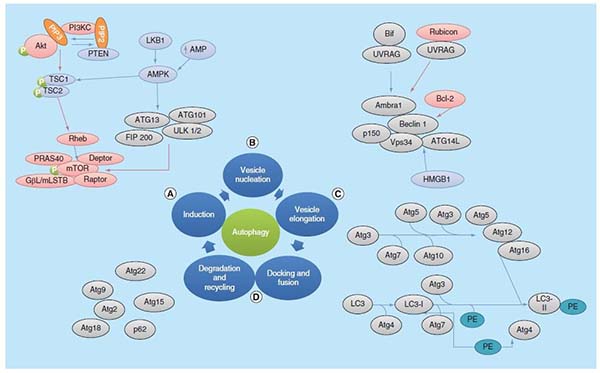
(1)Induction
Phagophore, a preautophagosomal structures assembly site, acts as the precursor for the formation of autophagosome. The initiation of autophagosome is regulated by ULK 1 complex. ULK1 complex consist of ULK1/2, Atg13, Atg 101 and focal adhesion kinase family interacting protein of 200 kDa (FIP200). mTORC1 is a protein complex that is inhibited by starvation and nutrient deprivation conditions. Activation of mTOR negatively regulates the autophagy. mTORC1 is made up of mTOR, regulatory associated protein of mTOR (raptor), GbL/mLST8, PRAS40 and DEPTOR. Under nutrient-rich conditions, mTOR is associated with this complex, and mTORC1 is activated by class I PI3K1/Akt signaling, resulting in the phosphorylation of ULK1/2 and Atg13, and prevention of phosphorylation of FIP200. Under nutrient deprivation conditions, inhibition of mTOR leads to its dissociation from the complex, which results in the dephosphorylation of Atg13 and ULK1/2, this facilitates ULK1/2 to phosphorylate FIP200 and thereby induces autophagy. Several signaling molecules regulate autophagy through mTOR. Insulin and insulinlike growth factor regulate via PI3K/Akt signaling and AMPK, which inhibits mTOR activity through Tsc1, Tsc2 and Rheb. AMPK is activated by LKB1 kinase. LKB1 kinase directly phosphorylate AMPK when cellular AMP/ATP ratio is increased(Figure 3A).
(2)Vesicle nucleation
This is a process in which proteins and lipids are recruited for construction of the autophagosomal membrane. This process is regulated by class III PI3K complex, consisting of Bif-1, Atg14L, Vps34, Vps15, UVRAG, Ambra1, Rubicon and Bcl-2/Bcl-XL [15]. Cytosolic HMGB1 is a proautophagic factor which binds to Beclin 1 and positively regulates the autophagy (Figure 3B).
(3)Vesicle elongation
Autophagosome elongation is controlled by two types of ubiquitylation-like reactions. In the first reaction, ATG5 and ATG12 are conjugated to each other in the presence of Atg7 and Atg10. Vps34 activity is essential for the formation of this conjugation. ATG5-ATG12 conjugate interacts with Atg16Ls producing the Atg16L complex, which localized to the growing autophagosomal membrane. In second ubiquitylation-like reaction, phosphatidylethanolamine (PE) and microtubule-associated protein1-light chain 3 (MAP1-LC3, also known as Atg8 and LC3) are conjugated. The conjugation of Atg5-Atg12 is believed to have role in conjugation of PE to LC3-I, which is generated by Atg4 through cleavage of the C-terminus of LC3. LC3-I covalently conjugates with PE and forms a membrane associated LC3II. The Atg5–Atg12–Atg16L complex targets LC3 for lipidation at phagophores. LC3II is the only protein that takes part in autophagosomes formation but not in other vesicular structures[15]. Autophagic flux is defined as the dynamic process of autophagosome synthesis, delivery of autophagic substrates to the lysosome and degradation of autophagic substrates inside the lysosome and is a more reliable indicator of autophagic activity (Figure 3C).
(4)Docking & fusion & degradation & recycling
(1)Oncogenes that regualte autophagy
Oncogenic autophagic pathways involving PI3KCI, Akt, mTORC, Ras, BCR–ABL, STAT, Bcl-2 and Bcl-XL have their own roles in cancer cell survival. The PI3K–Akt–mTOR signaling pathway regualtes autophagy and blockade of autophagy via the PI3K–Akt–mTOR pathway contributed to overcome chemotherapy resistance and resensitize the tumor cells to anticancer therapy. EGF as a key regulatory factor in cell survival can activate three signaling pathways that are necessary to the initiation and progression of cancer, Ras-MAPK, PI3K-Akt and the JAK-STATs. EGFR tyrosine kinase inhibitors can induce autophagy to promote tumor cell survival in response to targeted chemotherapies. Bcl-2 and Bcl-XL can also mediate autophagic pathway in cancer cells. Bcl-2 can block the interaction between tumor suppressive Beclin-1 and PI3KCIII/Vps34, thereby downregulating autophagic pathway. BCR–ABL can stimulate the transcription of mTORC1 via PI3K-Akt-FoxO signaling. Ras oncoproteins are member of family guanosine-5-triphosphates. Activation of Ras induces autophagic cell death or tumorigenesis which depends on extent of expression. Acute activation of Ras occurs in the absence of mutations (precancerous stage), resulting in cellular senescence or autophagic cell death via Beclin-1/Vps34 lipid kinase activation complex[16].
(2)Tumor suppressors that regualte autophagy
Tumor suppressive autophagic pathways contain some key regulators like Beclin-1, UVRAG, Bif-1, ATG5, p53 and FoxO1. Autophagy can be positively regulated by Beclin1 through combination with PI3KCIII/Vps34, and other positive and negative regulators such as Vps15, ATG14L/Barkor (Beclin 1-associated autophagy-related key regulator), UVRAG, Bif-1, Ambra1, Rubicon, HMGB1, Survivin, Akt and Bcl-2/Bcl-XL. Beclin-1 expression may reduce proliferation in cancer cells and decrease the tumorigenic potential which indicates its important role in autophagy mediated tumor suppression. Some antiapoptotic Bcl-2 family members, such as Bcl-2 and Bcl-XL, contain four Bcl-2 homology domains and inhibit autophagy through interacting with Beclin-1. Beclin-1 interaction can be blocked by Bcl-2. This decreases PI3KCIII activity and downregulates autophagy through either the disassociation of Beclin-1/PI3KCIII or inhibition of Beclin-1 activity. In addition, the suppression of ATG5, an essential autophagy gene, has a crucial inhibitory role in several types of human tumors. p53 is known as the guardian of the cellular genome. Mutations in p53 are most common as far as human cancers are concern. In response to DNA damage, oncogenic activation and hypoxia or other forms of stress p53 coordinates cellular responses, through both transcription dependent and independent mechanisms, to prevent or repair genomic damage or removes potentially oncogenic cells. p53 promotes or inhibits autophagy which depends on its localization. Cytoplasmic p53 promotes autophagic cell death and localization of p53 to the ER involves inhibition of autophagy. In starvation conditions p53 represses activation of autophagosomal membrane protein LC3 through a post-transcriptional mechanism and found to show cell survival function. In addition, cytoplasmic p53 can activate genes encoding component AMPKb1 of AMPK, SESN2, Tsc1 and Tsc2, and PTEN, which lead to inhibition of mTOR and, by association, to the activation of autophagy. p53 also regulates autophagy by interaction with other genes, damage-regulated autophagy modulator 1 DRAM1 and sestrin 1/2. DRAM1 encodes a lysosomal protein that induces autophagy. Sestrin-1 and sestrin-2 are two targets of DRAM, whose expressions are usually activated upon DNA damage and oxidative stresses, thus it may be p53-mediated tumor expression through an mTORC1 signaling[15].
Some autophagy activators such as perhexiline, niclosamide, amiodarone and rotterin have been identified to promote autophagy by inhibiting the function of mTORC1 in cancers under nutrient-rich conditions without blocking mTORC2[17].
The autophagic inducer, AZD8055, an ATP-competitive inhibitor of mTOR, can block phosphorylation of mTORC1 substrates as well as Akt in order to suppress tumor growth in mice by inducing autophagy. In lung cancer cells, sulindac sulfide amide, a N,N-dimethylethyl amine derivative of sulindac sulfide, found to have potent tumor cell growth-inhibitory activity by inhibiting Akt/mTOR signaling. Tricribine can induce autophagy through dephosphorylation of Akt and inhibition of mTORC1 in T-cell acute lymphoblastic leukemia. In human prostate cancer cells, phenethyl isothiocyanate(PEITC) can suppress the phosphorylation of both Akt and mTOR. Tetrahydrocannabinol(THC) induces cancer cell death through activation of autophagy via enhancing the ER stress that activates autophagy via inhibition of Akt and mTORC1[18].
Many types of neurodegenerative diseases are accompanied by the accumulation of aggregated and ubiquitinated proteins. Autophagy is involved in the degradation and removal of aggregated proteins, and the inhibition of constitutive autophagy leads to neurodegeneration in the central nervous system. The accumulation of autophagosomes in neurons is associated with neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease[19, 20]. Autophagic flux is controlled by the balance between autophagosome formation and autophagic degradation, the impairment of which causes neuronal cell death. Such accumulation indicates an imbalance between autophagosome formation and autophagic degradation. Excessive autophagy can lead to excessive degradation of cytosolic components and neuronal cell death.
(1)Alzheimer’s Disease
Alzheimer’s disease is the most common progressive neurodegenerative disorder and is characterized by dementia and morphological changes in the brain. The pathology of brains from individuals with Alzheimer’s disease indicates the amyloid plaques composed of accumulated amyloid-β(Aβ) peptides and the neurofibrillary tangles with tau. The autophagosome has secretase activity and is the place in which Aβ peptides are generated and accumulate in neurons from mouse models of Alzheimer’s disease. The excessive accumulation of autophagosomes in dystrophic neurites was observed in the brains of patients with Alzheimer’s disease. This indicates that there is a disruption in the degradation of cytosolic components in autolysosomes. Dysfunction of presenilin 1, which is associated with familial Alzheimer’s disease, causes the impairment of the acidification of lysosomes. The impairment of lysosomal acidification results in the inhibition of proteolysis in autolysosomes and the disruption of autolysosome formation, leading to the accumulation of autophagosomes and Aβ peptides in autophagosomes. Beclin1, which is necessary for the initiation of autophagy, has been shown to be reduced in the brains of patients with Alzheimer’s disease, and the reduction of beclin1 leads to decreased autophagy, the accumulation of Aβ peptides, and neurodegeneration in mouse models of Alzheimer’s disease[21].
(2)Parkinson’s Disease
Parkinson’s disease is the second-most common neurodegenerative disease after Alzheimer’s disease. Mutations in phosphatase and tensin homolog (PTEN)-induced putative kinase 1 (PINK1), which is encoded by the PARK6 gene, were identified in cases with early-onset Parkinson’s disease. Mutations in PINK1 or Parkin are the leading cause of parkinsonism. The dysfunction of PINK1 and Parkin in patients with Parkinson’s disease results in the impairment of mitophagy, which is regulated by NDP52 and OPTN[22, 23].
(3)Huntington’s Disease
Huntington’s disease is an autosomal-dominant neurodegenerative disease caused by a cytosine-adenine-guanine (CAG) expansion encoding a polyglutamine (polyQ) at the N-terminus of huntingtin (HTT) and is characterized by motor dysfunctions, cognitive disability, and psychiatric disturbance. Dysfunction of HTT results in neurodegeneration, indicating that HTT is essential for neurons. HTT also binds to p62, which links ubiquitinated substrates and LC3 to bring them to an autophagosome. It has been shown that the deletion of polyQ in HTT enhances neuronal autophagy. Thus, the conformational modification of HTT by polyQ expansion can restrain the autophagic pathways in the neurons and leads to neurodegeneration[24, 25].
Given that cancer represents approximately 200 different diseases, and autophagy has a global role in metabolism and protein and organelle quality control, it is not surprising that definition of the role of autophagy in cancer is still evolving. Autophagy stimulation may prevent disease, and although pharmacologic means to stimulate autophagy may have value, the same objective can be accomplished with periodic fasting. The potential effectiveness of this hypothesis is testable. Autophagy inhibition can compromise tumorigenesis, but the underlying mechanisms require definition. Highly selective and potent autophagy inhibitors are becoming available, but how to identify the patients most likely to respond is not yet clear. The means to measure autophagy activity in human tumors would be useful.

![[KO Validated] DNAJB1 Rabbit pAb](https://pic.abclonal.com.cn/A5504_P971_KO-WB_01.jpg?t=1767195828 "alt=")
![[KO Validated] DNAJB1 Rabbit pAb](https://pic.abclonal.com.cn/A5504_N21761-5_WB_02.jpg?t=1767195828 "alt=")
![[KO Validated] DNAJB1 Rabbit pAb](https://pic.abclonal.com.cn/A5504_N21761-5_WB_03.jpg?t=1767195828 "alt=")
![[KO Validated] DNAJB1 Rabbit pAb](https://pic.abclonal.com.cn/A5504_P972_IF_01.jpg?t=1767195828 "alt=")
![[KO Validated] DNAJB1 Rabbit pAb](https://pic.abclonal.com.cn/A5504_P972_IF_02.jpg?t=1767195828 "alt=")
![[KO Validated] DNAJB1 Rabbit pAb](https://pic.abclonal.com.cn/A5504_P972_IF_03.jpg?t=1767195828 "alt=")











_WB_01.jpg?t=1779441771 "alt=")













_WB_03.jpg?t=1765361541 "alt=")
_IF_01.jpg?t=1765361541 "alt=")
_IF_02.jpg?t=1765361541 "alt=")



